


Code-barres ADN pour caractériser la biodiversité
PDF
Les nouvelles techniques de biologie moléculaire haut-débit permettent d’identifier les espèces de notre environnement. En caractérisant de courts fragments de leur ADN qui persistent dans le milieu, il est ainsi possible d’inventorier la biodiversité d’un écosystème à partir d’échantillons d’eau, de sol ou d’excréments. L’intérêt de ces inventaires est extrêmement divers. Ils permettent de détecter la présence d’espèces discrètes, de reconstituer des paléo-environnements ou encore de caractériser des régimes alimentaires. Ils offrent aussi des perspectives pour la traçabilité par l’ADN et le contrôle d’authenticité des produits agro-alimentaires et cosmétiques.
1. Un outil de diagnostique universel
Caractériser la biodiversité en espèces est un enjeu scientifique et sociétal majeur. En effet, identifier les espèces est un préalable nécessaire à la compréhension de leurs interactions qui conditionnent le fonctionnement, la dynamique et l’évolution des écosystèmes. Cette étape est aussi nécessaire pour pouvoir conserver voire restaurer la biodiversité dans un contexte de changement global due aux variations du climat, au changement des modes d’utilisation des terres, à l’urbanisation. La description des espèces n’est pas une préoccupation nouvelle, elle concerne depuis toujours toutes des sociétés humaines qui prélèvent des organismes dans leur environnement à des fins diverses : alimentaires, médicales, culturelles, etc.
Les espèces ont été et sont encore décrites essentiellement sur la base de critères morphologiques, qui ont été complétés, au fil des progrès scientifiques, par des critères moléculaires reposant notamment sur la caractérisation de l’ADN (lire Qu’est-ce que la Biodiversité ?). Ces derniers sont particulièrement pertinents pour étudier des groupes dans lesquels la morphologie est difficilement accessible, comme les micro-organismes, ou peu variables, tels que les nématodes.
Le projet Barcode of Life [1] a démarré dans les années 2000 pour fournir un outil de diagnostic universel de la diversité, utilisable dans de nombreux domaines : écologie, agronomie, réglementation douanière, etc. Il a ainsi contribué à accélérer la description de la biodiversité encore inconnue. Ce projet repose sur la notion de code-barres ADN (en anglais DNA-barcoding) : il s’agit d’obtenir des données génétiques standardisées à partir de spécimens référencés dans des collections et identifiés par des taxonomistesPersonnes compétentes en taxonomie qui a pour objet de décrire les organismes vivants et de les regrouper en entités appelées taxons afin (a) de les identifier puis (b) les nommer et enfin les classer et (c) de les reconnaitre via des clés de détermination dichotomiques., pour identifier par la suite de façon fiable et rapide les espèces dont sont issus des individus prélevés dans leur environnement. Le code-barres ADN correspond au fragment standard d’ADN dont la séquence est caractéristique de l’espèce. Par exemple, pour caractériser des espèces animales, les chercheurs impliqués dans Barcode of Life ont défini une région du gène mitochondrial codant pour la cytochrome oxydase 1Sous-unité 1 du complexe enzymatique de la chaîne respiratoire (abréviation : COX1). Cette sous-unité est codée par le génome mitochondrial, contrairement à la majorité des gènes codant les sous-unités de la cytochrome oxydase (codés par le génome nucléaire). L’utilisation des séquences de COX1 permet de discriminer entre les diverses espèces animales, à l’exception des Cnidaires. comme fragment de référence.
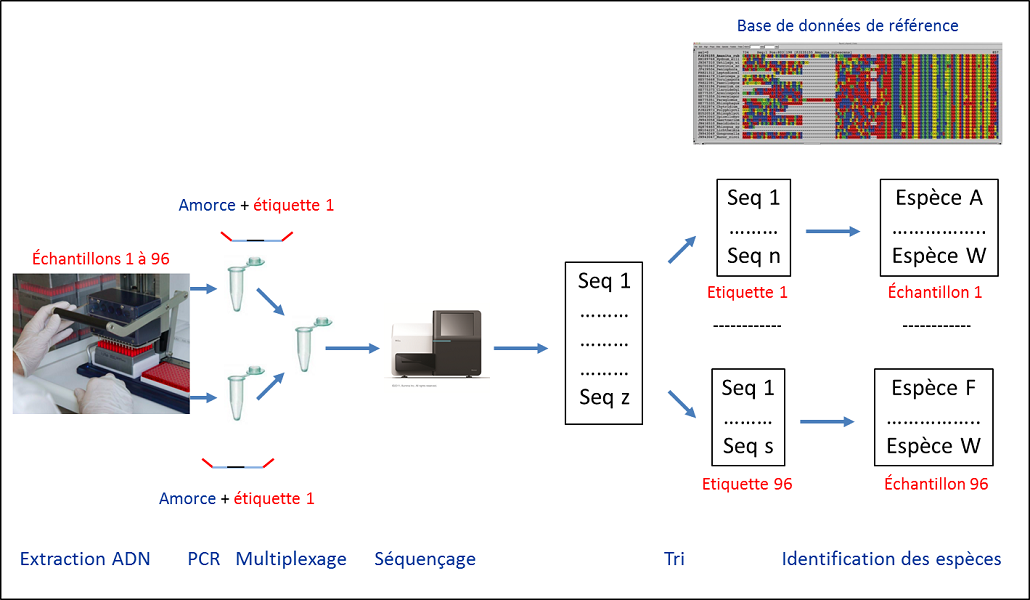
Cette notion de code-barres est utilisée par les taxinomistes, mais aussi par les écologistes qui la considère plus largement comme l’utilisation de toute technique d’analyse de l’ADN pour identifier des spécimens. Cette stratégie permet d’identifier les espèces présentes dans un milieu alors même que les individus n’y sont pas facilement accessibles. Cela concerne les micro-organismes bien sur, mais aussi les macro-organismes animaux et végétaux dont on peut détecter la présence grâce aux traces d’ADN qu’ils laissent : cadavres, mucus, excréments… Le principe ? Extraire l’ADN d’un échantillon environnemental (eau, sol, fèces), puis à amplifier par PCRAbréviation pour « Polymerase Chain Reaction » ou réaction en chaîne par polymérase. L’abréviation anglaise est passée dans le langage courant. Technique de réplication ciblée in vitro qui permet d’obtenir, à partir d’un échantillon complexe et peu abondant, d’importantes quantités d’un fragment d’ADN double brin, spécifique et de longueur définie. Chaque cycle de PCR est constitué de trois étapes : une dénaturation de l’ADN par chauffage pour séparer les deux brins qui le composent, une hybridation des amorces aux extrémités de la séquence recherchée, une élongation grâce à l’action d’une ADN polymérase. le fragment cible correspondant au code-barres à l’aide d’un couple d’amorcesSéquences oligonucléotidiques utilisées lors des réactions de PCR. Elles définissent, en la bornant, la séquence à amplifier. prédéfini [2]. Ces amorces peuvent être spécifiques d’une espèce. Cette approche permis de détecter la présence d’une espèce invasive comme la grenouille taureau, même à faible densité, à partir d’échantillons d’eau de mare. À l’inverse, les amorces peuvent être définies de manière pertinente pour étudier un large spectre d’espèces. On parle alors de metabarcoding. Dans ce cas, il faut séquencer les ampliconsFragments d’ADN amplifiés par PCR (réaction en chaîne par polymérase). produits par PCR, puis les comparer à une base de référence pour les relier à une espèce donnée (Figure 1).
2. Une approche de biologie moléculaire au service de l’écologie
Pour une approche de metabarcoding permettant de déterminer efficacement un ensemble d’espèces dont les ADN sont en mélange dans un échantillon environnemental, il est nécessaire de définir de bons codes-barres ADN. Ces code-barres doivent présenter une séquence variable entre espèces mais très conservée au sein d’une même espèce, afin d’avoir un fort pouvoir discriminant. Cette séquence doit être encadrée par deux zones très conservées d’une espèce à l’autre, pour permettre l’amplification du fragment par PCR chez le plus grand nombre d’espèces possible (vaste couverture taxonomique). La très bonne conservation des sites de fixation des amorces PCR est importante pour permettre une amplification non biaisée des espèces composant le mélange. De plus, le fragment amplifié doit être court pour permettre de caractériser des matrices dégradées. En effet, la dégradation fragmente l’ADN rendant difficile l’amplification de fragments de plus de 150 nucléotides. De plus, l’ADN étant généralement en quantité limitante, on privilégie l’utilisation de fragments d’ADN mitochondriaux ou chloroplastiques dont le nombre de copies par cellule est 100 à 1 000 fois supérieur à celui de l’ADN nucléaire. Il est également utile de définir des codes-barres ADN phylogénétiquement informatifs (i.e. dont le niveau de divergence reflète la divergence entre espèces) afin de pouvoir relier des espèces inconnues à des espèces connues proches. Les grandes bases de données, riches en séquences d’espèce variées sont maintenant utilisées à l’aide d’outils bio-informatiques dédiés pour définir le code-barres les plus pertinents pour étudier un groupe d’organismes. Il s’agit, à partir d’un jeu de séquences de référence prédéfini, de rechercher des zones courtes et variables encadrées par des zones conservées pour choisir les amorces d’amplification les mieux adaptées. Ces approches bio-informatiques permettent aussi d’estimer le pouvoir discriminant et la couverture taxonomique des codes-barres à partir de toutes les séquences théoriquement amplifiables dans une base de données.

- La première étape est l’échantillonnage ; il doit se faire selon des normes strictes évitant toute contamination (Figure 2).
- L’extraction de l’ADN de chaque échantillon est réalisée selon un protocole adapté au type d’échantillon à étudier (eau, sol, excrément…).
- Les ADN extraits sont ensuite amplifiés par PCR avec les amorces correspondant au code-barres. Par ailleurs, l’information nécessaire à l’attribution de chaque séquence à un échantillon donné doit impérativement être préservée. Cette information se présente sous la forme d’un oligonucléotide différent pour chaque échantillon. Il joue le rôle d’une étiquette (ou tag) placée à une extrémité des amorces d’amplification. Ces dernières sont identiques pour tous les échantillons (Figure 1). À cette étape, il est possible de bloquer l’amplification d’une espèce particulière en utilisant un oligonucléotide se positionnant spécifiquement sur l’ADN de l’espèce à exclure et qui empêche la réaction de polymérisation de l’ADN. Cette technique est, par exemple, utilisée pour étudier le régime alimentaire d’un carnivore à partir de fèces. Afin d’identifier toutes les proies potentielles, on utilise des amorces amplifiant l’ADN de tous les mammifères. Il faut cependant bloquer l’amplification de l’ADN du prédateur, présent en grande quantité et qui pourrait masquer la détection de certaines proies. Après PCR, chaque échantillon est alors représenté par un mélange des codes-barres ADN amplifiés (amplicons) des espèces qu’il contient.
- Les produits PCRProduits obtenus à partir de la réaction PCR. Ce sont des fragments d’ADN double brin. des divers échantillons à analyser sont mélangés au cours d’une étape de multiplexage réalisée avant séquençage (Figure 1).
- Le séquençage haut-débit des amplicons est réalisé. Le développement de ces nouvelles techniques a permis le développement des approches métabarcoding jusqu’alors inenvisageables dans la pratique. Les technologies actuelles permettent de séquencer en parallèle plusieurs millions de molécules d’ADN [3]. Partant de plusieurs centaines d’échantillons, quelques milliers de séquences sont suffisantes pour fournir une information pertinente.
- Après séquençage, les séquences sont triées par échantillon grâce aux tags, puis assignées à des espèces par comparaison avec des séquences de référence.
Tout au long de ce processus, l’outil bio-informatique est indispensable. Il permet de trier les données, de constituer les bases de référence, d’assigner les séquences aux taxonsUnités des classifications hiérarchiques des êtres vivants. Généralement le terme est employé aux rangs spécifique (l’espèce) et subspécifique (la sous-espèce). via ces bases, de définir des listes de tags et de gérer les erreurs de séquençage.
Sans aller jusqu’à l’identification des espèces, on peut utiliser les différentes séquences obtenues pour définir des unités opérationnelles (MOTUS pour Molecular Operationnal Taxonomic Units), à partir desquelles il est également possible de quantifier la biodiversité des échantillons. C’est généralement le cas lorsque l’on travaille sur des micro-organismes dont la plupart des espèces ne sont ni décrites ni cultivables.
3. Caractériser des échantillons environnementaux : l’approche métabarcoding
L’approche métabarcoding offre des alternatives aux méthodes souvent beaucoup plus lourdes à mettre en œuvre qui étaient jusqu’à présent utilisées pour décrire la biodiversité. Elle ouvre de nouvelles perspectives pour étudier le fonctionnement et l’évolution des écosystèmes, terrestres et aquatiques, qui requièrent la connaissance préalable des communautés d’espèces interagissant en leur sein [4]. Décrire la biodiversité à partir d’échantillons de terre en utilisant le métabarcoding se révèle, entre autres, utile lorsque les individus sont difficiles à trouver et à identifier morphologiquement. C’est le cas de nombreuses espèces de la faune du sol dont la fonction au sein de l’écosystème est essentielle : vers de terres, insectes, collemboles, etc. Le métabarcoding peut aussi se substituer aux relevés botaniques classiquement utilisés particulièrement dans des milieux où la diversité est extrêmement élevée comme les forêts tropicales. L’Amazonie renferme 11 000 espèces d’arbres dont la moitié présente un risque d’extinction. Les méthodes botaniques classiques conduiraient à ignorer jusqu’à 20 % des genres présents ; une bien meilleure résolution pouvant être apportée par l’utilisation de codes-barres ADN.
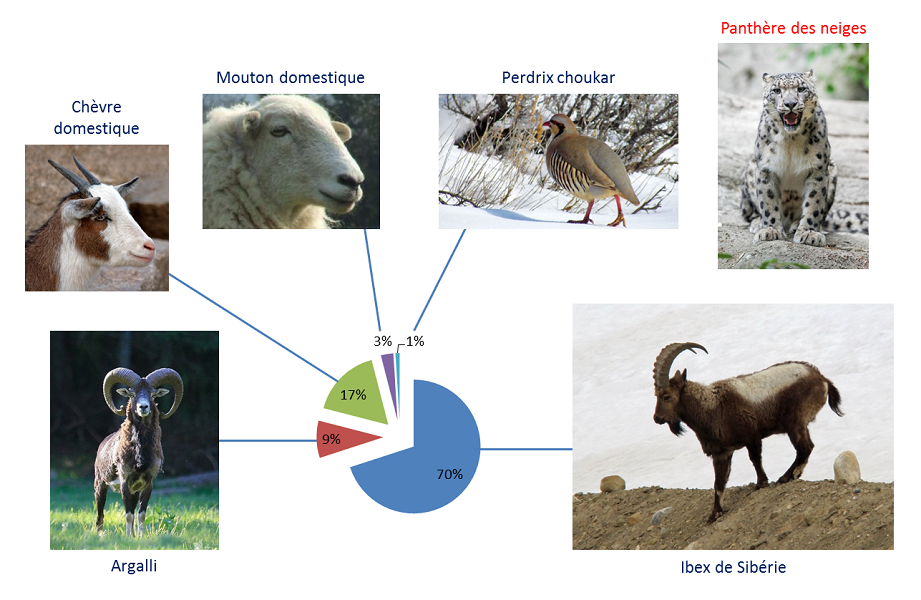
Enfin, le métabarcoding s’avère spécialement efficace pour reconstituer des paléo-écosystèmes alors même que les espèces qui les composaient ont disparu. Les méthodes classiquement utilisées, comme l’étude de macro fossiles et de pollens, sont lourdes à mettre en œuvre pour une faible résolution taxonomique. Des premières études de métabarcoding d’échantillons de permafrostTerme géologique qui désigne un sol dont la température se maintient en dessous de 0°C pendant plus de deux ans consécutifs. Représente plus de 20% de la surface terrestre de la planète. On parle de permafrost en anglais et de pergélisol en russe. Le permafrost est recouvert par une couche de terre, appelée « zone active », qui dégèle en été et permet ainsi le développement de la végétation. Son dégel sous l’effet du réchauffement climatique a des conséquences majeures pour l’environnement : relargage de méthane, libération de microorganismes pathogènes, etc. (lire Le permafrost) datant de plus de 20 000 ans ont démontré une bien meilleure résolution que les analyses des pollens des échantillons. L’étude de différentes couches de pergélisolsTerme utilisé à la place de permafrost pour la Sibérie. de Sibérie (voir Figure 2) a montré que les steppes de la période préglaciaire étaient constituées de plantes herbacées non graminoïdesPlantes herbacées, monocotylédones, majoritairement de la famille des Poaceae (graminées au sens strict), des Cyperaceae (laîches) et des Juncaceae (joncs). Elles ont une morphologie voisine du type « graminée », c’est-à-dire ayant notamment des tiges proches du « chaume », et des feuilles au limbe étroit et effilé. qui ont été remplacées par des graminées à la période postglaciaire. En parallèle, l’analyse de contenus d’estomacs fossilisés a montré que les mammouths, qui ont disparu après la glaciation, se nourrissaient principalement de ces plantes non graminoïdes. L’approche métabarcoding a ainsi permis de corréler le régime alimentaire des animaux disparus avec la variation drastique de la diversité végétale de leur écosystème.
Grâce au développement des nouvelles techniques de séquençage, le métabarcoding est devenu une alternative efficace aux méthodes traditionnelles pour décrire la biodiversité à partir d’échantillons environnementaux [6]. De plus, il offre de nouvelles perspectives pour réaliser des études intégrées à partir d’un même échantillon, via l’analyse combinée de différents codes-barres. On peut imaginer l’analyse simultanée du microbioteEnsemble des microorganismes (bactéries, levures, champignons, virus) vivant dans un environnement spécifique (appelé microbiome) chez un hôte (animal ou végétal). Un exemple important est l’ensemble des microorganismes vivant dans l’intestin ou microbiote intestinal, anciennement appelé « flore intestinale ». de l’intestin ou du rumen, du cortège de parasites et du régime alimentaire d’une espèce à partir de ses excréments. Les champs d’application ne se limitent pas aux études écologiques. Ils s’étendent aux domaines pour lesquels l’analyse de la composition de matrices complexes via la caractérisation de l’ADN est un enjeu : médecine légale, agro-alimentaire, contrôles douaniers…
Références et notes
Photo de couverture : © Jacques Joyard
[1] http://www.barcodeoflife.org
[2] http://www.ens-lyon.fr/RELIE/PCR/principe/principe.htm
[3] https://www.ebi.ac.uk/training/online/course/ebi-next-generation-sequencing-practical-course/what-you-will-learn/what-next-generation-dna-
[4] Pompanon F, Coissac E, Taberlet P (2011) Metabarcoding, une nouvelle façon d’analyser la biodiversité. Biofutur, 319:30-32.
[5] Shehzad W et al. (2012) Prey preference of snow leopard (Panthera uncia) in South Gobi, Mongolia. PLoS ONE 7(2): e32104. doi:10.1371/journal.pone.0032104
[6] Joly D, Faure D & Salamitou S (2015) Empreinte du vivant, l’ADN de l’environnement. Le Cherche Midi, 192 p.
L’Encyclopédie de l’environnement est publiée par l’Association des Encyclopédies de l’Environnement et de l’Énergie (www.a3e.fr), contractuellement liée à l’université Grenoble Alpes et à Grenoble INP, et parrainée par l’Académie des sciences.
Pour citer cet article : POMPANON François, SHEHZAD Wasim (3 février 2019), Code-barres ADN pour caractériser la biodiversité, Encyclopédie de l’Environnement. Consulté le 13 juin 2026 [en ligne ISSN 2555-0950] url : https://www.encyclopedie-environnement.org/vivant/metabarcoding-codes-barres-adn-caracteriser-biodiversite/.
Les articles de l’Encyclopédie de l'environnement sont mis à disposition selon les termes de la licence Creative Commons BY-NC-SA qui autorise la reproduction sous réserve de : citer la source, ne pas en faire une utilisation commerciale, partager des conditions initiales à l’identique, reproduire à chaque réutilisation ou distribution la mention de cette licence Creative Commons BY-NC-SA.


