Quand l’expédition Tara Océans explore la diversité du plancton
PDF
Les océans couvrent les deux tiers de notre planète, la vie y est née il y a plus de 3 milliards d’année. L’océan constitue une formidable pompe qui absorbe près de 30 % des émissions de dioxyde de carbone dues aux activités humaines, notamment grâce au phytoplancton qui le capte au cours de la photosynthèse. Le plancton, constitué d’organismes invisibles à l’œil nu, joue un rôle majeur dans les grands cycles biogéochimiques du carbone, de l’oxygène sur notre planète (la moitié de l’oxygène produit sur Terre provient des océans), de l’azote, du phosphore et du soufre. Pourtant, les micro-organismes qui participent à ces cycles nous restent pour l’essentiel encore inconnus. L’expédition Tara Océans a mis sur pied un consortium scientifique international pour développer des protocoles d’échantillonnage à haute résolution, analyser les échantillons, générer des données et les analyser pour explorer ce monde inconnu. Après trois ans de navigation et d’étude des océans planétaires, les chercheurs de l’expédition Tara Océans ont dévoilé une diversité insoupçonnée chez les organismes unicellulaires eucaryotes aussi appelés protistes. Le séquençage de près d’un milliard de codes-barres génétiques a permis de changer radicalement notre vision de la diversité biologique et fonctionnelle du plancton mondial, qui peuple un écosystème-clef pour le fonctionnement de notre biosphère.
1. Explorer le plus grand écosystème sur Terre

L’exploration de la distribution globale des communautés et diversités de microbes marins est devenue quantitative grâce au séquençage à haut débit disponible au milieu des années 2000, ce qui a ouvert la voie pour des campagnes d’échantillonnage à grande échelle spatiale. En effet, la distribution du plancton dépend fortement de facteurs abiotiques, comme la lumière, les nutriments, la turbulence, la température, la salinité ou le pH, et de facteurs biotiques, comme la présence d’autres organismes tels des prédateurs ou des symbiontes. Même si l’abondance locale du plancton varie de façon horizontale, verticale et saisonnière, les organismes planctoniques sont présents partout dans les océans.
L’importance du plancton à l’échelle planétaire est multiple :
- Il est à la base des chaînes alimentaires et représente 50% de la production annuelle de dioxygène sur Terre [1].
- Le métabolisme du plancton joue un rôle majeur dans les grands cycles biogéochimiques du carbone, de l’oxygène, de l’azote, du phosphore et du soufre.
- L’océan constitue aussi une formidable pompe qui absorbe près de 30 % des émissions de dioxyde de carbone (un gaz à effet de serre) dues aux activités humaines, notamment grâce au phytoplancton qui capte le dioxyde de carbone durant la photosynthèse (Lire : Un cycle du carbone perturbé par les activités humaines).
2. L’expédition Tara Océans

- Tara Arctic (2006-2008) a durant ses deux années de dérive sur la banquise a collecté des données relatives à l’atmosphère, la glace et l’océan ; Pris dans la glace pendant 504 jours et Tara a atteint la position la plus septentrionale jamais atteinte historiquement par un navire (à l’exclusion des brise-glaces) : 88°32’10 » N.
- Tara Océans (2009-2013) a permis de conduire une étude d’ampleur inédite sur le plancton, au cours d’un périple de 140 000 kilomètres sur tous les océans de la planète. L’expédition Tara Océans Polar Circle faisait également partie du projet Tara Océans, une mission sur 6 mois autour du Cercle Polaire et la première dans l’histoire à passer par les Passages Nord-Est et Nord-Ouest pendant la même année.
- Tara Méditerranée (2014) a permis d’évaluer l’impact des micro-plastiques sur la santé et le fonctionnement des écosystèmes en Méditerranée.
- Tara Pacific (2016-2018) a permis d’explorer les potentialités de résistance, d’adaptation et de résilience des récifs coralliens face au changement global.
- Tara Microplastiques (2019) a parcouru les 4 façades maritimes européennes et a prélevé des échantillons dans les 9 principaux fleuves d’Europe pour remonter aux origines de la pollution plastique.
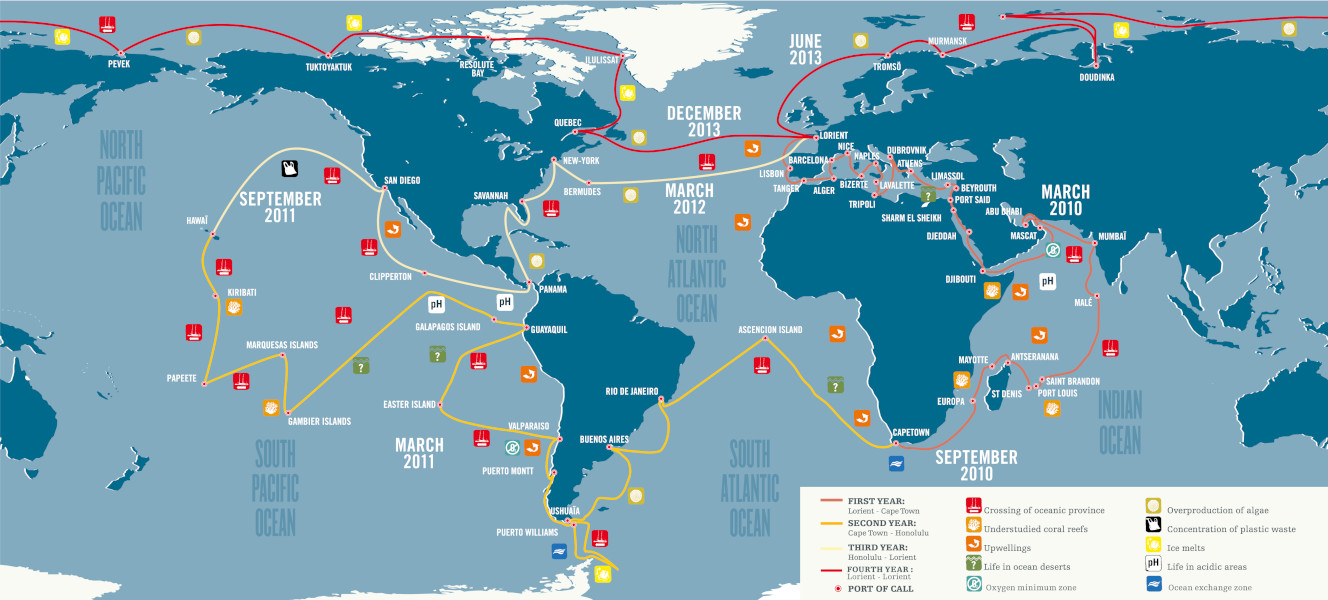
Au total, 40 000 échantillons d’eau de mer et de plancton ont été prélevés dans 210 stations réparties dans 20 provinces biogéographiques. De nombreuses questions ont animé cette expédition :
- Quelle est la vraie nature de la diversité planctonique dans nos océans ?
- Quels sont les organismes qui portent les fonctions les plus importantes ?
- Quel sont les effets des paramètres environnementaux et des interactions biotiques sur l’écosystème océanique ?
Afin de répondre à ces questions, l’expédition Tara Océans a regroupé plus de 250 scientifiques à travers le monde et procédé à un échantillonnage strictement identique pendant plus de trois ans sur la goélette longue de 36 mètres.
Le projet Tara Océans a utilisé de nombreuses nouvelles technologies et outils d’analyse pour établir le premier effort de collecte de données à l’échelle planétaire qui couple biogéographie, écologie, génétique et morphologie, regroupant une communauté internationale de scientifiques issus de disciplines bien différentes : écologistes marins, microbiologistes, océanographes, statisticiens, biogéochimistes, informaticiens, ingénieurs.
Le programme d’échantillonnage standard a été conçu pour étudier une grande variété d’écosystèmes marins : remontées d’eau (upwellings), points chauds de biodiversité, zones de bas pH ou pauvres en oxygène… Un total de 210 stations a été défini (Figure 3) sur lesquelles une caractérisation environnementale plus précise a été conduite, afin de contextualiser les prélèvements morphologiques et génétiques du plancton.
3. Grands principes d’échantillonnage
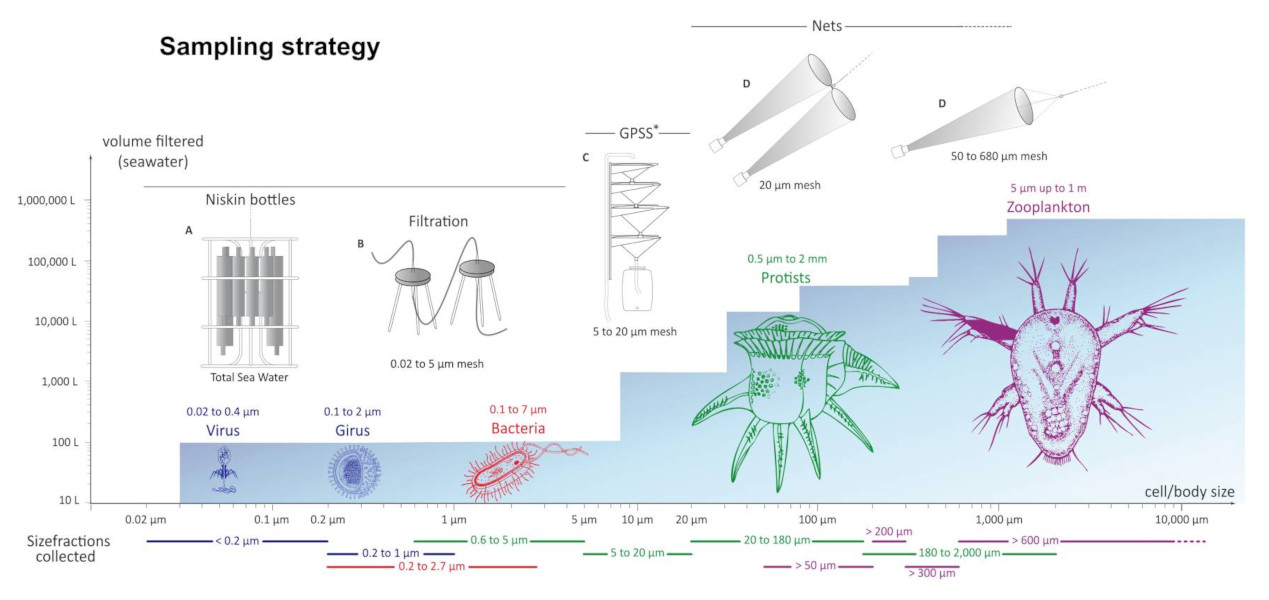
- La première est la couche d’eau de surface (SUR), définie comme la couche de trois à sept mètres sous la surface.
- La seconde est la couche dite « deep chlorophyll maximum » (DCM) qui correspond à la zone d’abondance maximale du plancton photosynthétique, déterminée grâce à la mesure de la concentration en chlorophylle par fluorimétrie. L’existence de ce maximum s’explique par un compromis entre les deux conditions nécessaires à la croissance du phytoplancton : présence de lumière et apport de nutriments par les eaux froides de profondeur.
- Le troisième échantillonnage a été effectué dans la zone dite « mésopélagique », en-dessous de la DCM et où la lumière depuis la surface ne passe plus, se situant typiquement à une profondeur de 700 mètres.
Pour chaque station, l’échantillonnage s’est fait sur plusieurs fractions de taille d’organismes (Figure 4) [2].
Le plancton prélevé pendant l’expédition Tara Océans couvre six ordres de grandeur en termes de taille qui correspondent aux :
- virus, virus géants (giant virus ou girus) (Lire Focus Virus des océans);
- procaryotes (bactéries et archées) ;
- eucaryotes unicellulaires (protistes) ;
- eucaryotes pluricellulaires (comme les copépodes).
Les cellules des protistes mesurent entre 0,8 et 2000 microns. Des filets de tailles de maille appropriées ont été utilisés afin de créer plusieurs fractions de tailles : 0,8-5 microns, 5-20 microns, 20-180 microns, 180-2000 microns.
Vidéo 1 : Tara Océans (avec photo ci-dessous): prélèvement du plancton. Filtration du plancton (GPSS).
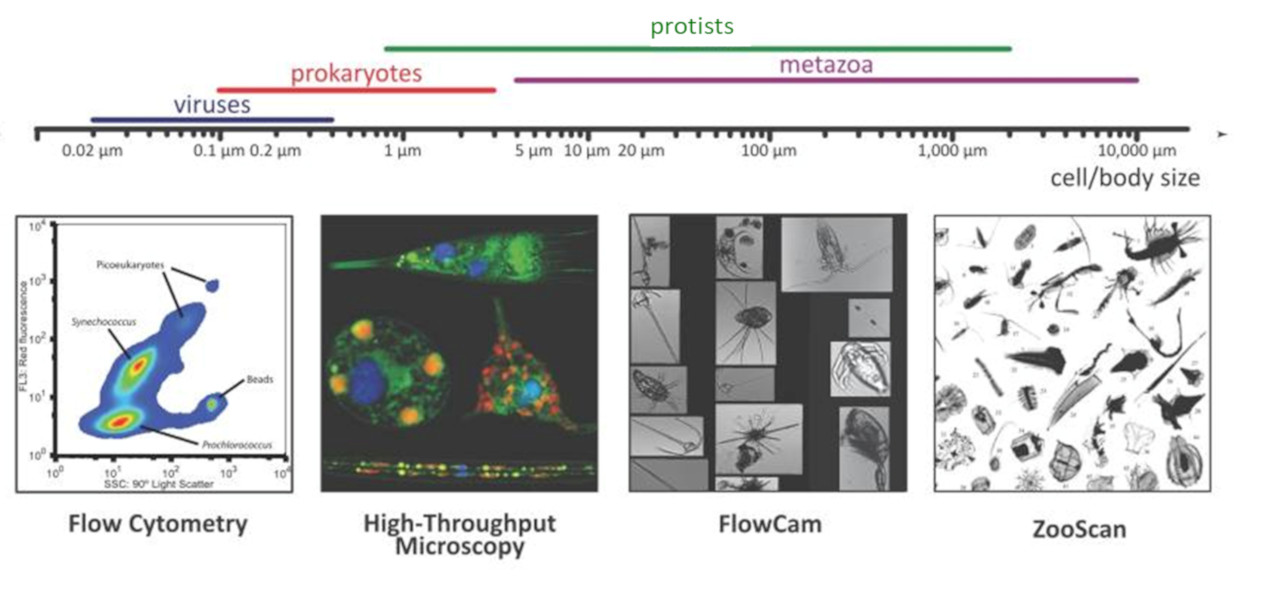
- D’une part, des systèmes de reconnaissance automatisés, tels que le FlowCam [3] et le ZooScan [4], ont permis des mesures quantitatives de la biodiversité d’organismes allant de 20 microns à quelques centimètres.
- D’autre part, la microscopie confocale 3D et la microscopie électronique à transmission ont permis des analyses ultra-structurales détaillées des petits protistes.
4. Analyse de la diversité génétique du plancton
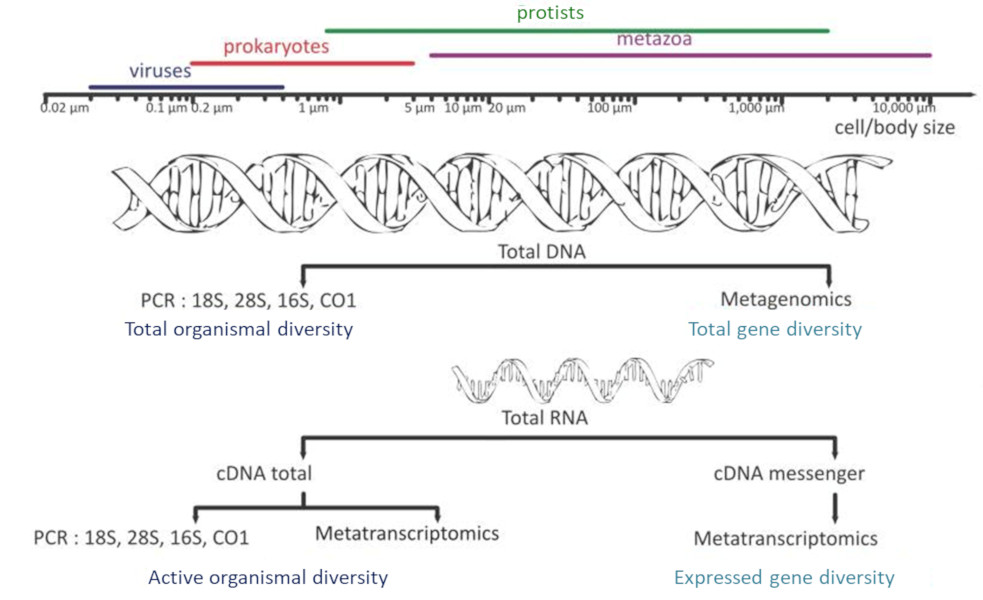
- Séquençage de l’ADNr 18S (metabarcoding et PCR), le gène codant l’ARN ribosomique 18S : Qui est là ?
- Métatranscriptomique : détermination de l’ensemble des ARN produits – le transcriptome – par les différents organismes collectés lors du processus de transcription d’un gène en protéine : Qui fait quoi ?
- Métagénomique : caractérisation de l’ensemble des génomes présents dans l’échantillon environnemental : Qui peut faire quoi ?
Le préfixe « meta » ajouté aux termes « barcoding », « transcriptomique » et « génomique » exprime l’idée que ces analyses moléculaires sont réalisées non pas sur une seule espèce mais sur un ensemble d’espèces provenant d’échantillons environnementaux. Le séquençage ADN à haut débit [5],[6] a ouvert la porte à l’exploration, à la fois qualitative et semi-quantitative, de la diversité génétique d’échantillons environnementaux.
4.1. Qui est là ?
L’exploration de la diversité génétique des échantillons s’est basée sur l’utilisation de différentes technologies moléculaires.
Barcoding moléculaire (DNA barcoding). La phylogénie des microorganismes a longtemps été fondée sur des caractères morphologiques et biochimiques. Récemment, les marqueurs moléculaires (codes-barres ou barcodes) ont été utilisés pour reconstruire l’histoire évolutive des organismes vivants, en se basant sur l’idée – simplifiée – que plus deux organismes sont distants évolutivement, plus la différence entre leur séquence génomique sera grande.
Stricto sensu, un code-barre ADN est une courte séquence (typiquement 100 à 400 paires de bases) correspondant à une portion standard du génome (par exemple l’ADN ribosomal 18S). Il peut être utilisé pour identifier les espèces, comme un code-barre utilisé au supermarché qui permet de faire la relation entre le produit (la séquence) et son prix (l’identification de l’espèce) (Lire : Code-barres ADN pour caractériser la biodiversité). Cette séquence est choisie sur la base de critères précis [7] :
- la variabilité intra-espèce doit être faible: cette séquence doit être quasi identique chez tous les organismes de la même espèce ;
- la variabilité inter-espèces doit être grande afin que l’on puisse différencier deux espèces différentes sur la base de leur séquence.
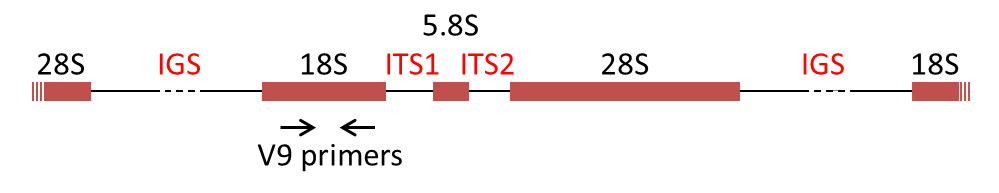
Les marqueurs moléculaires ADN les plus utilisés pour la reconstruction phylogénétique sont les gènes codant les ARNr des sous-unités ribosomales, les ADNr. Chez la majorité des eucaryotes, l’ARNr 18S est présent dans la petite sous-unité ribosomale, tandis que la grande sous-unité contient trois molécules d’ARNr (5S, 5,8S, 28S chez les mammifères et 25S chez les plantes). Les gènes codant les ARNr sont souvent regroupés en batterie (cluster), et séparés par des espaceurs internes transcrits (internal transcribed spacers, ITS1 et ITS2) et un espaceur intergénique (IGS) (Figure 7) [8].
Metabarcoding. L’impact des codes-barres comme outils moléculaires va bien au-delà d’une phylogénie à plus grande résolution des espèces connues : conservation, découverte d’espèces, et écologie des communautés en bénéficient [9],[10]. Grâce à l’avènement du séquençage à haut débit, le barcoding moléculaire (DNA barcoding) est devenu un outil répandu pour l’écologie des communautés eucaryotes (et procaryotes !) à travers une méthode connue sous le nom de metabarcoding.
Plus formellement, le metabarcoding ADN fait référence à l’identification automatisée de plusieurs espèces provenant d’un même échantillon contenant les organismes entiers, ou d’un échantillon environnemental contenant de l’ADN dégradé (du sol, de l’eau, fèces, etc.) [11]. Un bon code-barre pour une étude de metabarcoding moléculaire (comme l’ADNr 18S) doit :
(i) correspondre à une portion de gènes quasi identiques au sein des individus de la même espèce, mais qui diffèrent entre espèces ;
(ii) être utilisable pour toutes les espèces considérées dans l’étude ;
(iii) permettre l’assignation taxonomique et ce, à différents niveaux taxonomiques [6].
Une fois extrait, l’ADN de l’échantillon est amplifié par PCR (polymerase chain reaction ; amplification en chaîne par polymérase). La PCR est une technique permettant de dupliquer en grand nombre (avec un facteur de multiplication de l’ordre du milliard) une séquence spécifique d’ADN (ou d’ARN) à partir :
- d’une faible quantité (de l’ordre de quelques picogrammes) d’acide nucléique ;
- d’amorces spécifiques nucléotidiques dites « universelles », comme la séquence appelée V9 [12], ce qui permet d’amplifier spécifiquement la portion d’ADN choisie (l’amplicon, par exemple, l’ADNr 18S) de tous les organismes présents.
Le produit PCR final obtenu après amplification est donc un mélange de toutes les séquences V9 des organismes dans l’échantillon analysé.
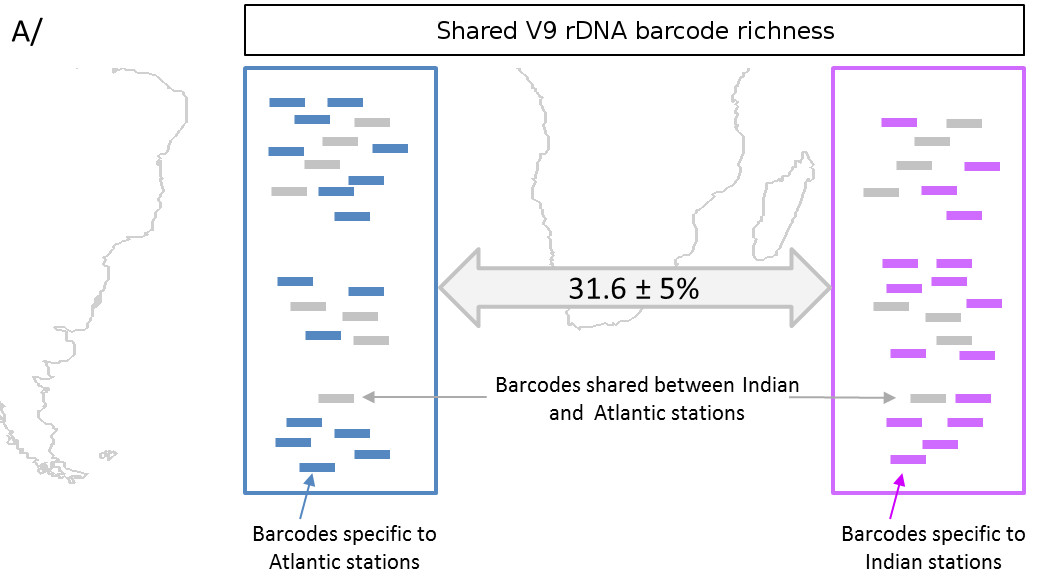
Cette assignation taxonomique se fait en comparant les UTO de l’échantillon avec des bases de données d’organismes de référence. L’idée est de voir de quelle séquence connue l’UTO se rapproche le plus, et dans quelle proportion. Le degré de similarité permettra de donner avec un certain degré de certitude un nom d’espèce, ou de genre, ou de famille : si une UTO a une séquence 100% identique à une espèce de nom connu, on lui attribue l’identité de cette espèce.
Mais parfois les séquences ne sont pas assez similaires, on s’arrêtera donc au niveau du genre et non de l’espèce.
Seul hic, plus de 40% des UTO déterminées dans le cadre de l’expédition Tara Océans sont totalement nouvelles et ne correspondent à aucune espèce connue. On sait simplement qu’il s’agit d’Eucaryotes mais il est quasiment impossible de les placer dans un arbre phylogénétique. Excitant !
4.2. Qui fait quoi ?
![]()
Pour étudier spécifiquement les transcrits eucaryotes, il est possible de sélectionner uniquement les ARNm spécifiques des eucaryotes (Figure 9) [16],[17]. Ces ARNm sont ensuite rétrotranscrits en présence d’une transcriptase inverse : les ADN complémentaires obtenus sont séquencés et leurs séquences comparées à celles présentes dans les bases de données de référence, ce qui peut permettre à la fois de préciser une annotation fonctionnelle (de quels gènes ces transcrits proviennent-ils, à quoi peuvent-ils servir ?) et taxonomique (de quels organismes ces transcrits proviennent-ils ?). C’est le « qui fait quoi », détaillé dans Carradec et al. [17].
4.3. Qui peut faire quoi ?
Aujourd’hui, et principalement pour le domaine procaryote, les analyses génomiques portent sur les séquençages de génomes entiers, ou métagénomique.[18] Au lieu d’utiliser des amorces PCR spécifiques (voir plus haut Metabarcoding), l’ensemble des génomes dans l’échantillon environnemental est alors séquencé.
- La première étape, après extraction de l’ADN, consiste à fragmenter tous les ADNs présents dans l’échantillon en morceaux très courts puis à les séquencer ; on parle de « shotgun sequencing » ou séquençage aléatoire.
- Par la suite, les fragments séquencés sont assemblés bioinformatiquement à partir des régions chevauchantes, afin de reconstruire les génomes d’origine.
Le séquençage « métagénomique » rend ainsi compte de l’ensemble des gènes non pas exprimés, mais présents dans l’échantillon. Ce type de séquençage informe sur le potentiel génétique de la communauté d’organismes de l’échantillon, c’est le « qui peut faire quoi », mais ne le fait pas forcément. Cette approche au potentiel colossal se confronte à de nombreuses limitations techniques.
À l’époque de l’expédition Tara Océans, la taille des fragments d’ADN pouvant être séquencés à haut débit dépassait à peine les 500 paires de base. Si l’on considère par exemple le génome d’une diatomée (phytoplancton abondant), qui fait au moins 30 millions de paires de bases, cela signifie qu’il faut séquencer au minimum 60 000 fragments d’ADN pour reconstituer ce génome complet (Figure 10). En pratique, les analyses ne sont jamais réalisées sur un seul individu, mais à partir d’un mélange de fragments d’ADN provenant d’individus différents.
En réalité, il n’est pas possible de se limiter au séquençage de 60 000 fragments au hasard, car sinon certaines parties du génome de notre diatomée auront été séquencées plusieurs fois, mais d’autres aucune fois, et le génome reconstitué comportera donc des « trous ». Il faut en outre disposer de fragments d’ADN qui se chevauchent pour pouvoir reconstituer l’ordre des séquences.
Pour pallier ce problème, on séquence l’équivalent non pas d’un seul génome (60 000 fragments) mais, par exemple, de vingt génomes (soit 1 200 000 fragments !).
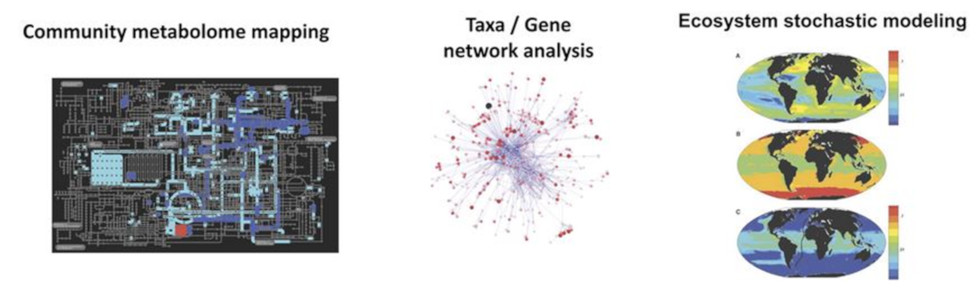
Couplées à l’imagerie quantitative, ces approches de séquençage à haut débit permettent non seulement de comprendre la structure, la localisation et la fonction des gènes, mais aussi et surtout d’explorer la diversité du plancton. Grâce à des méthodes puissantes reposant sur la bioinformatique, ces analyses apportent des données sur l’évolution, le métabolisme et les interactions entre les organismes (Lire : Focus La pompe à carbone biologique de l’océan), et permettent de reconstruire les métabolomes de la communauté, des réseaux de gènes et d’organismes, ainsi que des modèles de distribution spatiale d’espèces (Figure 11).
5. Une diversité insoupçonnée : les protistes du plancton
-
- Présentation des résultats de Tara Océans à la Maison des Océans Tara Océans
- Présentation des résultats de Tara Océans à la Maison des Océans Tara Océans
Dans cette étude, les chercheurs ont déchiffré et analysé près d’un milliard de séquences d’ADN ribosomique, issues de 46 sites d’échantillonnage seulement. Ces séquences sont utilisées comme marqueurs de la biodiversité des eucaryotes (Lire : Qu’est-ce que la biodiversité ? et La biodiversité n’est pas un luxe mais une nécessité), des plus petits organismes unicellulaires (<1 micron) aux animaux planctoniques de quelques millimètres [22].
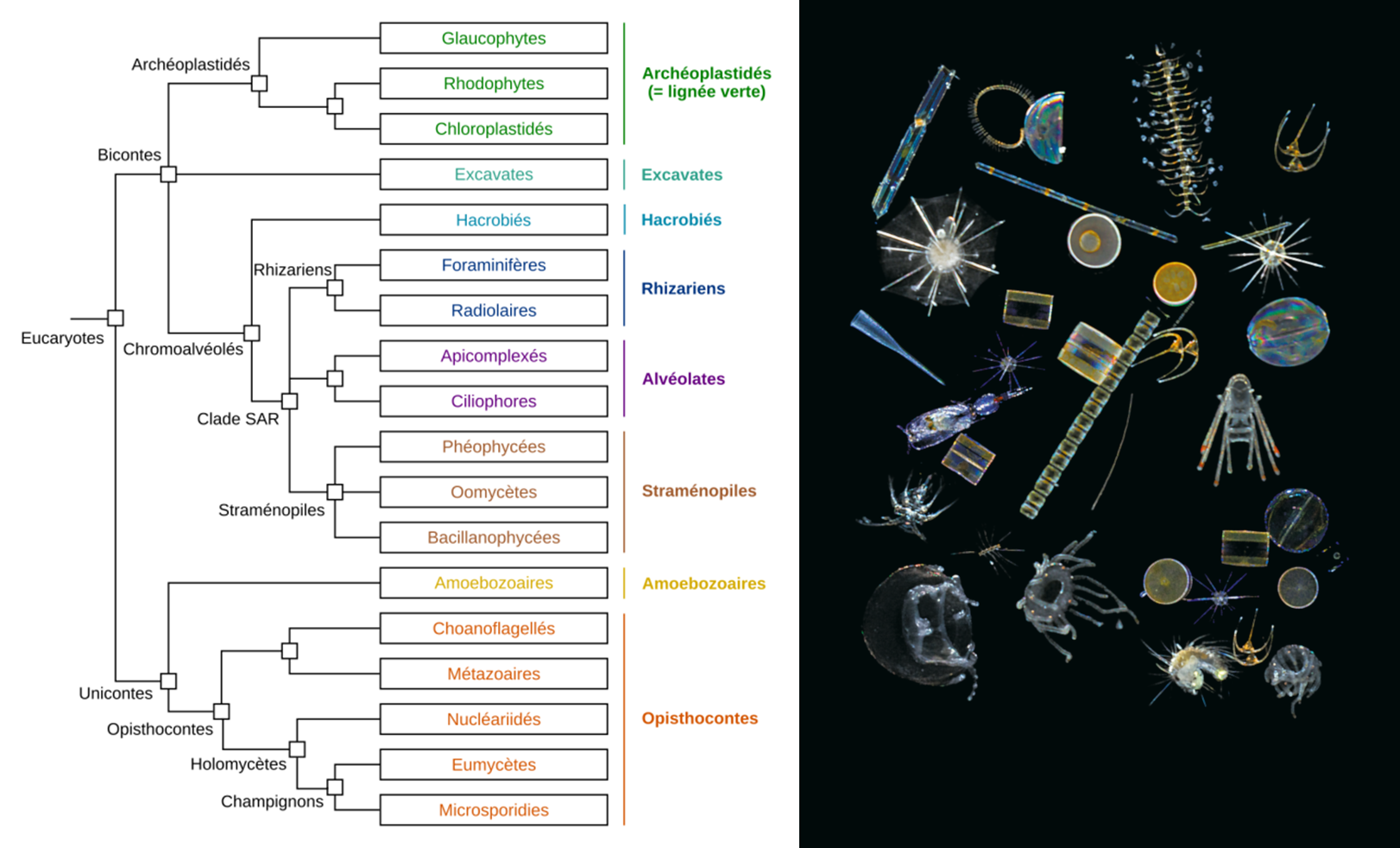
La grande quantité de codes-barres génétiques générés a tout d’abord permis de caractériser la quasi-totalité des espèces eucaryotes du plancton de la zone photique analysée. 150 000 types génétiques de plancton eucaryote ont été mis en évidence, ce qui représente une diversité insoupçonnée par rapport aux 11 000 espèces décrites jusqu’à présent. Il est apparu que la grande majorité des types génétiques répertoriés n’a pas de référent proche dans les bases de données génétiques actuelles, démontrant que ces organismes sont pour la plupart non-répertoriés et non cultivables. Un tiers de la diversité génétique n’a pu être associé à aucune des grandes lignées eucaryotes reconnues aujourd’hui.
Parmi les types génétiques pouvant être classés dans l’arbre de la vie eucaryote, la plupart se sont avérés correspondre à des organismes unicellulaires ou protistes, avec une diversité phénoménale de parasites, d’espèces symbiotiques, et de prédateurs en tout genre. Les organismes photosynthétiques étaient, quant à eux, bien moins diversifiés, plus petits, et représenteraient une biomasse largement plus faible.
À ce jour, plus de 100 publications émanent directement de l’analyse des données générées par l’expédition Tara Océans, dont cinq articles fondateurs ayant fait l’objet d’un numéro spécial « Tara Oceans » de la revue Science en 2015 (interrogation de la base de données ISI Web of Science sur la période 2009-2019 en utilisant les seuls termes « Tara Oceans » ; 2 Octobre 2020).
6. Messages à retenir
- Après trois ans de navigation autour du globe, les chercheurs de l’expédition Tara Océans ont réalisé une étude du plancton la plus exhaustive possible et permis de réaliser une photographie d’ensemble de l’écosystème planctonique mondial
- Ces travaux ont permis de découvrir 150 millions de gènes marins, 100 000 espèces et de déterminer le facteur de répartition des communautés planctoniques grâce à plus de 40 000 échantillons d’eau de mer. Ils ont en particulier dévoilé une diversité insoupçonnée chez les protistes (organismes unicellulaires eucaryotes).
- Plus de 100 publications scientifiques dans des revues de premier plan émanent directement de l’analyse des données générées par l’expédition Tara Océans. Ces données sont librement accessibles à tous les scientifiques, quelle que soit leur communauté.
- En parallèle de ces activités de recherche, le projet Tara Océans a visé aussi à sensibiliser le grand public aux problématiques liées au changement climatique, à travers de nombreux ateliers, visites à bord, outils pédagogiques disponibles sur leur site et validés par des scientifiques.
- La science ainsi générée a été aussi mise au service de la gouvernance climatique : en tant qu’observateur spécial à l’ONU, l’expédition Tara Océans a mobilisé les décideurs politiques au plus haut niveau.
Organisation scientifique initiale de Tara Océans :
Eric Karsenti, EMBL, Directeur scientifique, Etienne Bourgois Directeur de Tara Expéditions, Romain Troublé Secrétaire général de Tara Expéditions – Stephanie Kandels Lewis (EMBL) & Didier Velayoudon (DVIP consulting) Managers opérationnels
Coordinateurs scientifiques de Tara Océans : Ce groupe interdisciplinaire et international a élaboré la stratégie d’échantillonnage et le plan d’ensemble, fixé les objectifs généraux de l’expédition et du projet, dirigé l’expédition et effectué la majeure partie de l’analyse des résultats au cours des dix dernières années. Voir référence [23]
Notes et références
Image de couverture. [Source : Photo © G.Bounaud_C.Sardet_La Niak_Fondation Tara Océan]
[1] Field CB. 1998. Primary production of the biosphere: integrating terrestrial and oceanic components, Science 281, 237–240.
[2] Des échantillons d’eau préservés grâce à du paraformaldéhyde ont été utilisés pour la microscopie à haute résolution.
[3] FlowCam https://www.embrc-france.fr/fr/prestation/flowcam
[4] ZooScan https://www.embrc-france.fr/fr/prestation/zooscan
[5] L’ADN et l’ARN du plancton récolté lors de l’expédition Tara Océans ont été décryptés au Centre national de séquençage – Genoscope. Créé en 1996 pour participer au projet Génome humain et développer des programmes de génomique en France. Il a ensuite mis le cap vers la génomique environnementale et la métagénomique.
[6] Dardel F. & Képès F., 2002, Bioinformatique : génomique et post-génomique, Éditions de l’École Polytechnique 153-180 p. (ISBN 978-2-7302-0927-4, lire en ligne).
[7] Valentini A, Pompanon F, Taberlet P. 2009. DNA barcoding for ecologists, Trends in Ecology & Evolution, 24, 110–117.
[8] Zagoskin M, Lazareva V, Grishanin A & Mukha D. 2014. Phylogenetic information content of Copepoda ribosomal DNA repeat units: ITS1 and ITS2 impact. BioMed Research International, 926342. https://doi.org/10.1155/2014/926342
[9] Kress WJ, Erickson DL, Uriarte M & Garcı C. 2015. DNA barcodes for ecology, evolution, and conservation. Trends in Ecology & Evolution, 30, 25-35.
[10] Bucklin A, Lindeque PK, Rodriguez-Ezpeleta N, Albaina A & Lehtiniemi M. 2016. Metabarcoding of marine zooplankton: prospects, progress and pitfalls. Journal of Plankton Research, 38, 393–400.
[11] Taberlet P, Coissac E, Pompanon F, Brochmann C & Willerslev E. 2012. Towards next generation biodiversity assessment using DNA metabarcoding, Molecular Ecology, 21, 2045–2050.
[12] En comparant les séquences déjà connues d’ADNr 18S entre divers organismes, une région appelée V9 a été identifiée. Elle est longue de 130 paires de bases et ses extrémités sont très conservées chez tous les organismes connus. Les extrémités de cette séquence peuvent donc servir de point d’ancrage pour les amorces durant la PCR.
[13] Villar E. et al. (2015) Environmental characteristics of Agulhas rings affect interocean plankton transport. Science DOI:10.1126/science.1261447
[14] Blaxter M, Mann J, Chapman T, Thomas F, Whitton C., Floyd R & Eyualem A. 2005. Defining operational taxonomic units using DNA barcode data. Philosophical Transactions of the Royal Society B: Biological Sciences, 360, 1935–1943.
[15] L’ARN messager ou ARNm est une copie transitoire d’une portion de l’ADN correspondant à un ou plusieurs gènes. L’ARNm est utilisé comme intermédiaire par les cellules pour la synthèse des protéines. Dans une cellule, la population d’ARNm correspond aux gènes exprimés et sera ensuite traduite en protéines.
[16] Ce sont des ARNm dits polyadénylés : ils présentent dans la partie 3’ d’une queue faite d’une succession d’adénosines, absente chez les procaryotes (voir ref [6]).
[17] Carradec Q. et al. 2018. A global ocean atlas of eukaryotic genes. Nature Communications 9, 1038.
[18] Sacha Schutz Introduction à la métagénomique
[19] L’assemblage consiste à aligner et/ou fusionner des fragments d’ADN ou d’ARN issus d’une plus longue séquence afin de reconstruire la séquence originale, grâce à des outils bioinformatiques. Le problème de l’assemblage peut être comparé à celui de la reconstruction du texte d’un livre à partir de plusieurs copies de celui-ci, préalablement déchiquetées en petits morceaux.
[20] L’annotation d’un génome consiste à analyser la séquence nucléotidique qui constitue l’information brute pour en extraire l’information biologique. Elle permet d’une part de localiser les gènes et les régions codantes et d’autre part d’identifier ou de prédire leur fonction biologique (c’est l’annotation fonctionnelle). Ces deux étapes reposent initialement sur l’utilisation d’outils algorithmiques sophistiqués, dont le développement constitue l’un des champs de la bio-informatique.
[21] Lecointre G & Le Guyader H. 2016. Classification phylogénétique du vivant – Tome 1, Éditions Belin, Collection Nature, 584 pp.
[22] de Vargas C, Audic S, Henry N, Decelle J, Mahe F, Logares R, Lara E, Berney C, Le Bescot N, Probert I, Carmichael M, Poulain J, Romac S, Colin S, Aury JM, Bittner L, Chaffron S, Dunthorn M, Engelen S, Flegontova O, Guidi L, Horak A, Jaillon O, Lima-Mendez G, Lukes J, Malviya S, Morard R, Mulot M, Scalco E, Siano R, Vincent F, Zingone A, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Acinas SG, Bork P, Bowler C, Gorsky G, Grimsley N, Hingamp P, Iudicone D, Not F, Ogata H, Pesant S, Raes J, Sieracki ME, Speich S, Stemmann L, Sunagawa S, Weissenbach J, Wincker P & Karsenti E. 2015. Eukaryotic plankton diversity in the sunlit ocean. Science, 348, 1261605.
[23] Coordinateurs scientifiques de Tara Oceans :
Silvia Acinas, Department of Marine Biology and Oceanography, Institut de Ciències del Mar (CSIC), Barcelona, Catalonia, Spain.
Peer Bork, Structural and Computational Biology, European Molecular Biology Laboratory, Meyerhofstr. 1, 69117 Heidelberg, Germany.
Emmanuel Boss, School of Marine Sciences, University of Maine, Orono, Maine 04469, USA.
Chris Bowler, PSL Research University, Institut de Biologie de l’Ecole Normale Supérieure (IBENS), CNRS UMR 8197, INSERM U1024, 46 rue d’Ulm, F-75005 Paris, France.
Colomban de Vargas, CNRS, UMR 7144, EPEP & Sorbonne Universités, UPMC Université Paris 06, Station Biologique de Roscoff, 29680 Roscoff, France.
Mick Follows, Department of Earth, Atmospheric and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, MA, USA.
Gaby Gorsky, Sorbonne, UPMC Université Paris 06, CNRS, Laboratoire d’oceanographie de Villefranche (LOV), Observatoire Océanologique, 06230 Villefranche-sur-Mer, France.
Nigel Grimsley, CNRS, UMR 7232, BIOM, Avenue du Fontaulé, 66650 Banyuls-sur-Mer, France.
Pascal Hingamp, Aix Marseille Univ, Université de Toulon, CNRS, IRD, MIO, Marseille, France.
Daniele Iudicone, Stazione Zoologica Anton Dohrn, Villa Comunale, 80121 Naples, Italy.
Olivier Jaillon, Genoscope, Institut de biologie François Jacob, Commissariat à l’Energie Atomique (CEA), CNRS, Université Evry, Université Paris-Saclay, Evry, France
Lee Karp-Boss, School of Marine Sciences, University of Maine, Orono, Maine 04469, USA.
Uros Krkic, Cell Biology and Biophysics, European Molecular Biology Laboratory, Meyerhofstrasse 1, 69117 Heidelberg, Germany.
Fabrice Not, CNRS, UMR 7144, Sorbonne Universités, UPMC Université Paris 06, Station Biologique de Roscoff, 29680 Roscoff, France.
Hiroyuki Ogata, Institute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto, 611-001, Japan.
Stephane Pesant, MARUM, Center for Marine Environmental Sciences, University of Bremen, Bremen, Germany.
Jeroen Raes, Department of Microbiology and Immunology, Rega Institute, KU Leuven, Herestraat 49, 3000 Leuven, Belgium.
Emmanuel Reynaud, Earth Institute, University College Dublin, Dublin, Ireland.
Christian Sardet, Sorbonne Université UPMC Université Paris 06, CNRS, Laboratoire d’oceanographie de Villefranche (LOV), Observatoire Océanologique, 06230 Villefranche-sur-Mer, France.
Mike Sieracki, National Science Foundation, Arlington, VA 22230, USA.
Sabrina Speich, Laboratoire de Physique des Océans, UBO-IUEM, Place Copernic, 29820 Plouzané, France.
Lars Stemmann, Sorbonne, UPMC Université Paris 06, CNRS, Laboratoire d’oceanographie de Villefranche (LOV), Observatoire Océanologique, 06230 Villefranche-sur-Mer, France.
Matthew Sullivan, Department of Microbiology, The Ohio State University, Columbus, OH 43214, USA.
Shini Sunagawa, Structural and Computational Biology, European Molecular Biology Laboratory, Meyerhofstr. 1, 69117 Heidelberg, Germany.
Jean Weissenbach, Genoscope, Institut de biologie François Jacob, Commissariat à l’Energie Atomique (CEA), CNRS, Université Evry, Université Paris-Saclay, Evry, France
Patrick Wincker, Genoscope, Institut de biologie François Jacob, Commissariat à l’Energie Atomique (CEA), CNRS, Université Evry, Université Paris-Saclay, Evry, France
L’Encyclopédie de l’environnement est publiée par l’Association des Encyclopédies de l’Environnement et de l’Énergie (www.a3e.fr), contractuellement liée à l’université Grenoble Alpes et à Grenoble INP, et parrainée par l’Académie des sciences.
Pour citer cet article : VINCENT Flora, BOWLER Chris (4 février 2026), Quand l’expédition Tara Océans explore la diversité du plancton, Encyclopédie de l’Environnement. Consulté le 14 juillet 2026 [en ligne ISSN 2555-0950] url : https://www.encyclopedie-environnement.org/vivant/expedition-tara-oceans-plancton/.
Les articles de l’Encyclopédie de l'environnement sont mis à disposition selon les termes de la licence Creative Commons BY-NC-SA qui autorise la reproduction sous réserve de : citer la source, ne pas en faire une utilisation commerciale, partager des conditions initiales à l’identique, reproduire à chaque réutilisation ou distribution la mention de cette licence Creative Commons BY-NC-SA.